
(Journal of Food Science and Technology Nepal. 2: 128-131)
Upendra Thapa Shreshtha(1), Gyan Sundar Sahukhal(1), Subarna Pokhrel(1), Kiran Babu Tiwari(1), Anjana Singh(2), Vishwanath Prasad Agrawal(1*)
(1) Research Laboratory for Agricultural Biotechnology and Biochemistry (RLABB), Universal Science College, Maitidevi, Kathmandu, Nepal;
(2) Central Department of Microbiology, Tribhuvan University, Kirtipur, Nepal
*Address for Correspondence: Prof. Dr. Vishwanath Prasad Agrawal, RLABB, Tel: +977-1-4442775, E-mail: vpa@wlink.com.np
ABSTRACT
Bacillus thuringiensis strains were isolated from soil samples collected from Khumbu Base Camp of the Everest region and characterized by standard methods. Crystal protein (d-endotoxin) was extracted from the crystal protein producing strains (46 from Phereche and 40 from Sagarmatha National Park) from stationary phase culture broth and tested for insect bioassay. Crystal proteins were further purified by Native-PAGE. Among ten randomly selected isolates, one isolate showed the highest insecticidal activity against Dipteron insects. Its crystal protein had molecular weight about 120-130KD (revealed by SDS-PAGE) and was used to produce polyclonal antibody in New Zealand’s white rabbits. The presence of polyclonal antibody was confirmed by Ouchterlony double diffusion method. Indirect ELISA was optimized coating 6-8mg crystal protein per well in microtitre plate. The optimal dilution of the polyclonal antibody was 1000 folds corresponding to OD450 = 0.045 for color observation. Of the total 86 crystal protein producing isolates, crystal proteins from 31 isolates (36.05%) were 25-30% crossreactive, two groups of 6 isolates (6.97%) were 75-80% and 85-90% crossreactive respectively, and 4 isolates (4.65%) were 80-85% crossreactive with the polyclonal antisera. Only 3 isolates (3.49%) were more than 90% crossreactive. The Discriminatory Index (D) of the Indirect ELISA was 0.92.
Keywords: Khumbu region, d-endotoxin, Crystal protein, Insect-bioassay, Immunodiffusion, Immunocrossreactivity.
INTRODUCTION
Microbial insecticides are especially valuable as their toxicity to non target animals and humans is extremely low compared to other commonly used chemical insecticides. They are safer for both the pesticide user and consumers of pesticide treated crops (Neppl, 2000). The soil bacterium Bacillus thuringiensis fulfills the requisites of a microbiological control agent against agricultural pest and vectors that cause massive crop destruction (Ben-Dov et al, 1999). The main target pest of B. thuringiensis insecticides include various Lepidoptera (butterfly), Diptera (fliesand mosquitoes), and individual Coleopteran (Beatle) species and some strains kill off nematodes (Schnepf et al , 1998) where as B. thuringiensis var. kurstaki HD1 is highly potent strain due to its wide spread insecticidal properties (Dulmage, 1970).
Insect bioassay and rocket immunoelectrophoresis are currently used to detect and measure the levels of crystal proteins. Though the rocket immunoelectrophoresis is more sensitive than insect bioassay, it requires a considerable amount of antigen, at least 10 mg/ml (Wie et al, 1982). Immunodiffusion and ELISA are more practical and reliable methods. ELISA measures changes in enzyme activities proportional to the antigen or antibody concentrations. It is a highly versatile and sensitive analytical procedure for qualitative and quantitative determination of antibodies and almost any kind of antigens. The method discriminates different epitopes very efficiently provided that antibodies of high specificity and affinity are available. The detection limits of the assay may be well below 1 ng/ml (Perlmann and Perlmann, 2001). Hence ELISA can be effectively used to study cross reactivity of a given type of antigen. Identical antigens possess 100% crossreactivity with the given antisera and non identical ones don’t show any degree of crossreactivity. Thus the diversity of the given antigen and hence organisms in a given complex population can be studied by their cross reactivities. In order to study crystal protein diversity of B. thuringiensis strains, Indirect ELISA procedure was optimized in this study.
METHODOLOGY
Soil sampling, isolation and biochemical characterization: Soil samples were collected from Sagarmatha National Park (SNP) and Phereche of Khumbu Base Camp of Everest region and were transported to RLABB, where the study was carried out from March 2005 to December 2005 in joint collaboration with Central Department of Microbiology, Tribhuvan University, Kirtipur, Nepal. Bacillus thuringiensis were isolated by acetate selection method (Travers et al, 1987). The isolated organisms were identified by standard microbiological techniques including colonial and morphological characteristics, and biochemical tests (Bergey’s Manual, 1986).
Collection of Mosquito larvae: Mosquito (Lepidoptera) Larvae were collected from the ditches in local area of Bode, Bhaktapur, Nepal for insect bioassay. The larvae were identified as Culex spp. by zoologists at the Central Department of Zoology, Tribhuvan University, Kirtipur and bioassay was performed as described by Pang (1994).
Extraction and purification of crystal proteins: B. thuringiensis strains were incubated in Brain Heart Infusion broth (Calf brain infusion 200 g/l, beef heart infusion 250 g/l, protease peptone 10 g/l, dextrose 2 g/l, sodium chloride 5 g/l, disodium phosphate 2.5 g/l, final pH 7.4 ±0.2 at 25°C) and sterilized by autoclaving (15 lbs pressure 121oC, 15 min) at 30°C for 3-7 days till autolysis. Spores and crystals were separated by centrifugation (10,000 rpm, 20 min, 4 oC), and then washed four times with phosphate buffer (pH 7, 0.05M). The pellet was finally suspended overnight in carbonate buffer of pH 10.5 (0.05 M sodium carbonate, 0.01M b-mercaptoethanol, 1mM EDTA, 1mM PMSF) with constant shaking at 23-26oC, and centrifuged (10000 rpm, 20 min, 4 oC). Protein content in the supernatant was determined by Bradford assay (1976). In order to determine which proteins are responsible for the biological activities, they were electrophoresed under non denaturing conditions by Native PAGE (Blackshear, 1984) and the major bands were sliced , grinded in a minimum volume of phosphate-buffered saline (10 mM sodium phosphate, 150 mM NaCl, pH 7.2) , centrifuged (10000 rpm, 20 min, 4 oC) and concentrated using 20% TCA.
Insect bioassay and Molecular weight determination: For insect bioassay 10 larvae were taken in a jar containing 100 ml of sterilized water containing 0.3 ml of 5% Brewer's Yeast. Five ml of B. thuringiensis stationary phase culture was added and allowed to stand for 3 days. The number of deaths was recorded for one, two and three days. For the purified crystal protein bioassay, proteins (30µg/ml per assay) from each band were tested for insecticidal property as done by Pang (1994). Proteins from insect bioassay positive band was electrophoresed to determine molecular weight using molecular weight markers (lysozyme 14 KD, casein 22 KD, BSA 66KD) according to themethod of Laemmli (1970).
Polyclonal antiserum production: Proteins from insect bioassay positive band was resuspended in saline to a concentration of 1.0 mg/ml. The suspension was emulsified in an equal volume of Freund’s complete adjuvant (Difco, USA). A pair of New Zealand’s white rabbit was injected with 500 µg of the emulsified proteins (1.25 ml) by the intramuscular route in hind limbs. The booster injections with incomplete adjuvant were given three times in 14 days interval. The animals were bled 7 days after third booster dose. Polyclonal antiserum was pooled and decomplemented by incubation at 56°C for 30 min. Aliquots of antiserum (0.1 to 0.5 ml) were stored at -20°C until assayed.
Immunodiffusion and ELISA: The presence of polyclonal antibody was confirmed in a 1% agarose gel (0.05 M phosphate buffer, pH 7.2) by the Ouchterlony method (Talwar and Gupta, 1997) where 50 µl undiluted and diluted (1:10,1:100, 1:1000,1:10000 dilutions) antisera was poured in a centre well surrounded by 5 wells in petri-plate. 50 µl crystal protein antigen preparations (1 mg/ml) from SNP and Phereche isolates were applied in the surrounding wells against antiserum. The petri-plate was incubated at 4oC for 72 hours in a moist chamber. Immuno crossreactivity of B. thuriengensis crystal proteins was studied by indirect ELISA, where optimal dilutions of polyclonal antiserum and its second antibody (anti rabbit IgG conjugated with Horse Radish Peroxidase, Sigma, USA) was determined by chequerboard titration method (Trottier et al, 1972; Voller et al, 1976). To coat 96-well polystyrene microtitre plate, 100 µl crystal protein antigen (6µg) prepared in PBS (137 mM NaCl, 1.76 mM KH2PO4, 8.1 mM Na2HPO4, 2.7 mM KCl; pH 7.2 ) was applied in each well, and incubated overnight at 4oC (Trottier et al, 1972).Uncoated protein was washed 3 times using washing buffer (0.05%Tween 20 in PBS). For blocking, 200µl of 2% BSA in PBS was applied in each well and allowed to stand for 1 hour at room temperature. Blocking was done twice. After 3 times washing, each well was incubated with 100 µl polyclonal antibody of different dilutions for 2 hours at room temperature, and unbound polyclonal antibody was washed 4 times. The microtitre plate was then incubated with 100 µl of second antibody of different dilutions per well at room temperature for 1 hour and washed 4 times. Finally it was incubated with 100 µl tetramethyl benzidine substrate solution (0.01% tetramethyl benzidine in citrate phosphate buffer of pH 4.9, 2µl of 30% H2O2), reaction was allowed proceed for half an hour, stopped adding 1N HCl and the intensity of color was read at 450 nm within 10 min in ELISA reader (Dynatech MR-250).
Calculation of discriminatory index value (D): D value was calculated according to the formula of Hunter and Gaston (1988).Where, N=Numbers of isolates;S=Number of different polymorphic types.
RESULTS AND DISCUSSION
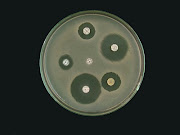
Total 109 B. thuringiensis isolates were obtained from soil samples of Phereche and SNP. Sixty three and 46 isolates were obtained from 52 Phereche and 39 SNP soil samples respectively. All the isolates were Gram positive rods. Of the 109 total isolates, only 86 isolates (79.63%) were positive on crystal protein staining, which were further proceeded for immunological characterization (Bergey’s Manual, 1986).Molecular weight determinationFive bands were observed for S6 preparation having molecular weight of 40, 58, 71, 83 and 107 KD in SDS-PAGE and a single band (Mol. Wt. -120-130KD) was observed in Native PAGE. This indicates that S6 crystal protein is composed of 5 different protein subunits. Similar result was observed by Drobniewski and Ellar (1989) and Pfannenstiel et al (1986) in their work on B. thuringiensis.Insect bioassayThe result of insect bioassay of crystal protein from SNP and Phereche B. thuriengensis isolates is displayed in Figure 1. For insect bioassay, cultures were grown to stationary phase which is suitable for the sporulation and production of crystal protein (Hunter and Gaston, 1988), and were used for the bioassay on mosquito larvae. Insecticidal activity of S6 endotoxin (30µg/ml) partially purified from autolysed B. thuringiensis broth by alkaline solution method following purification in Native PAGE is found to be 100 % (10/10) efficient, and hence it was used for polyclonal antibody production. Purification by Native PAGE has also been reported by Pang (1994). These crystal proteins are solubilized in the alkaline environment of Lepidoptera larval midgut, and then processing by midgut proteases results in a relatively stable, mature toxin (Van Rie et al., 1990). Activated cry toxins have two known functions, receptor binding and ion channel activity. The activated toxin binds readily to specific receptors on the apical brush border of the midgut microvilli of susceptible insects. Binding is a two-stage process involvingreversible and irreversible steps. The latter steps may involve a tight binding between the toxin and receptor, insertion of the toxin into the apical membrane, or both. It has been generally assumed that irreversible binding is exclusively associated with membrane insertion. Soon after insertion, toxins made pores on membrane and enter to body system. As soon as they enter to body they stop feeding and alternately death of insects occurs due to blood poisoning (Schnepf et al, 1998).Immunodiffusion and ELISADouble immunodiffusion performed in agarose gel gives clearly visible precipitin bands against crystal proteins of B. thuringiensis isolates from both SNP and Phereche upto 1:100 dilution of polyclonal antisera tested.(Fig. 2). The method gives reproducible results to detect crystal protein antigen with 100% specificity. For performing ELISA (Fig. 3), first and second antibody of dilutions 1:1000 and 1:2000 were optimal respectively. Below OD450 = 0.045, there was not clearly visible change in color to naked eyes. Trottier et al (1992) chose the optimal dilution of first and second antibodies difference between ELISA value with and without antigens with all other conditions remaining same. Between each ELISA step, plates were washed five times with a microtitre plate washer in order to prevent the false positive result (Trottier et al, 1992). The discriminatory index value, D (Table 1), was 0.92 (N=86, S=22). Hence the Indirect ELISA method divided all isolates into 13 groups with 92% confidence (Table-1). In which only 3 isolates (3.49%) were above 90% cross-reactive. The result clearly shows that B. thuringiensis isolates from Khumbu Base Camp are highly diverse for their crystal protein antigenicity. The S6 isolate showing potent insecticidal property tested against lepidopteron insects need to be studied further in larger trials so that it can have applicability to reduce the massive crop yield loss in this region.
CONCLUSION:
B. thuringiensis is diversed into 13 different groups in Khumbu Base Camp of Mount Everest region. Crystal protein from one of the most potent strain (S6) is 100% effective against lepidopteron insects.
ACKNOWLEDGEMENT
We express our full gratitude to CNR (Italy’s National Research Council) for supporting this work and especially thank to Mr. Yogan Khatri, Mr. Deepak Singh and Rajendra Aryal for collecting soil samples from Mount Everest region. Finally my (Upendra Thapa Shrestha) special regards goes to my dear parents for their everlasting support.
REFERENCES
Ben-Dov E, Wang Q, Zaritsky A, Manasherob R, Barak Z, Schneider B, Khamraev A, Baizhanov M, Glupov V, Margalith Y. Multiplex PCR screening to detect cry9 genes in Bacillus thuringiensis strains. Appl Environ Microbiol 1999; 65: 3714-6.
Bergey’s Manual of Systematic Bacteriology, Volume 2, 1986).Pang AS. Production of antibodies against Bacillus thuringiensis delta-endotoxin by injecting its plasmids. Biochem Biophys Res Commun 1994; 202: 1227-34.
Blackshear PJ (1984). Systems for polyacrylamide gel electrophoresis. In Methods in enzymology (Jakoby WB eds.) vol 104: 237-255.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248-54.
Drobniewski FA and Ellar DJ. Purification and properties of a 28-kilodalton hemolytic and mosquitocidal protein toxin of Bacillus thuringiensis subsp. darmstadiensis 73-E10-2. J Bacteriol 1989;171: 3060-7.
Dulmage HT Production of spore-delta-endotoxin complex by variants of Bacillus thuringiensis in two fermentation media. J Invertebr Pathol 1970; 16: 385-9.
Hunter PR and Gaston MA. Numerical Index of the Discriminatory Ability of Typing Systems: an Application of Simpson's Index of Diversity. Journal of Clinical microbiology 1988; 26: 2465-6.
Laemmli, UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970; 227:680-5.Laemmli, UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970; 227:680-5.
Neppl CC (2000). Managing Resistance to Bacillus thuringiensis Toxins. Environmental Studies University of Chicago.
Perlmann P and Perlmann H (2001) Enzyme-Linked Immunosorbent Assay ENCYCLOPEDIA OF LIFE SCIENCES 2001 Nature Publishing Group.Travers RS, Martin PA and Reichelderfer CF Selective Process for Efficient Isolation of Soil Bacillus spp. Appl Environ Microbiol 1987; 53: 1263-6.
Pfannenstiel MA, Couche GA, Ross EJ and Nickerson KW. Immunological relationships among proteins making up the Bacillus thuringiensis subsp. israelensis crystalline toxin. Appl Environ Microbiol 1986;52: 644-9.
Schnepf E, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, Zeigler DR and Dean DH. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev 1998; 62: 775-806.
Talwar GP and Gupta SK (1997). A hand book of practical and clinical immunology, second edition, volume 1, CBS publishers and distributors, Daryaganj, New Delhi.
Trottier YL, Wright PF and Lariviere S Optimization and standardization of an Enzyme-Linked Immunosorbent Assay protocol for serodiagnosis of Actinobacillus pleuropneumoniae serotype 5. J Clin Microboil 1992;30(1):46-53.
Van Rie, J., S. Jansens, H. Ho¨fte, D. Degheele, and H. Van Mellaert. Receptors on the brush border membrane of the insect midgut as determinants of the specificity of Bacillus thuringiensis delta-endotoxins. Appl. Environ. Microbiol 1990;56:1378-85.
Voller A., Bidwell D and Bartlett A. (1976) Microplate enzyme immunoassays for the immunodiagnosis of virus infections Manual of clinical immunology. American Society for Microbiology p. 506-512.
Wie SI, Andrews R JR, Hammock B, Faust R.M and Bulla LA () Enzyme-Linked Immunosorbent Assays for Detection and Quantitation of the Entomocidal Parasporal Crystalline Protein of Bacillus thuringiensis subspp. kurstaki and israelensist Appl Environ Microbiol: 1982, 891-4.
















{kind=link}